单细胞转座酶可及性染色质测序(scATAC-seq)技术的飞速发展为在单细胞水平研究基因调控机制的提供了全新契机。然而,现有scATAC-seq的分析流程往往依赖于伪批量(pseudo-bulk)峰调用(peak calling)策略,这不仅在一定程度上削弱了分析结果的单细胞分辨率,还极易掩盖罕见细胞类型的独特染色质特征,限制了对细胞异质性的精细刻画。
针对上述问题,清华大学信息国家研究中心/自动化系张学工团队在Nature Communications期刊上发表了题为“A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis”的研究论文。该研究系统构建了通用染色质可及性参考集cPeak,填补了传统单细胞分析中缺乏统一特征参考的空白,不仅在细胞精准注释和罕见细胞类型识别上展现出超越现有方法的卓越性能,还揭示了不同性质的染色质潜在可及区域在细胞分化与肿瘤进展中的动态变化规律。该成果为scATAC-seq数据分析提供了如同转录组分析中参考基因组般的统一特征坐标体系,极大地提升了跨数据集分析的精度和可靠性,并为构建染色质可及性细胞图谱、开发基于染色质可及性的基础大模型奠定了重要技术基础。

文章发表在Nature Communications
研究团队通过分析证实,基因组上潜在的染色质可及性区域在不同条件下表现出相对保守的区域和高度一致的“形状”特征(图1),提示这些区域可能是基因组的一种固有属性。基于这一观察,研究团队提出为染色质可及性数据构建一个类似于转录组分析中“参考基因组”的标准化特征集合。为此,研究团队系统整合了涵盖多种人体组织器官的624个高质量bulk ATAC-seq公共数据集,定义了约140万个高置信度的共识峰(consensus Peak, cPeak)。基于潜在染色质可及区域位置与其DNA序列特征相关的假设,团队进一步开发了卷积神经网络(CNN)深度学习模型,额外预测约28万个全新的cPeak。系统评估表明,cPeak在不同组织器官、测序平台上表现出极高的形状一致性和优异的真实数据覆盖率;同时,基因组注释结果也清晰地揭示了cPeak与关键基因调控元件的紧密关联(图1)。
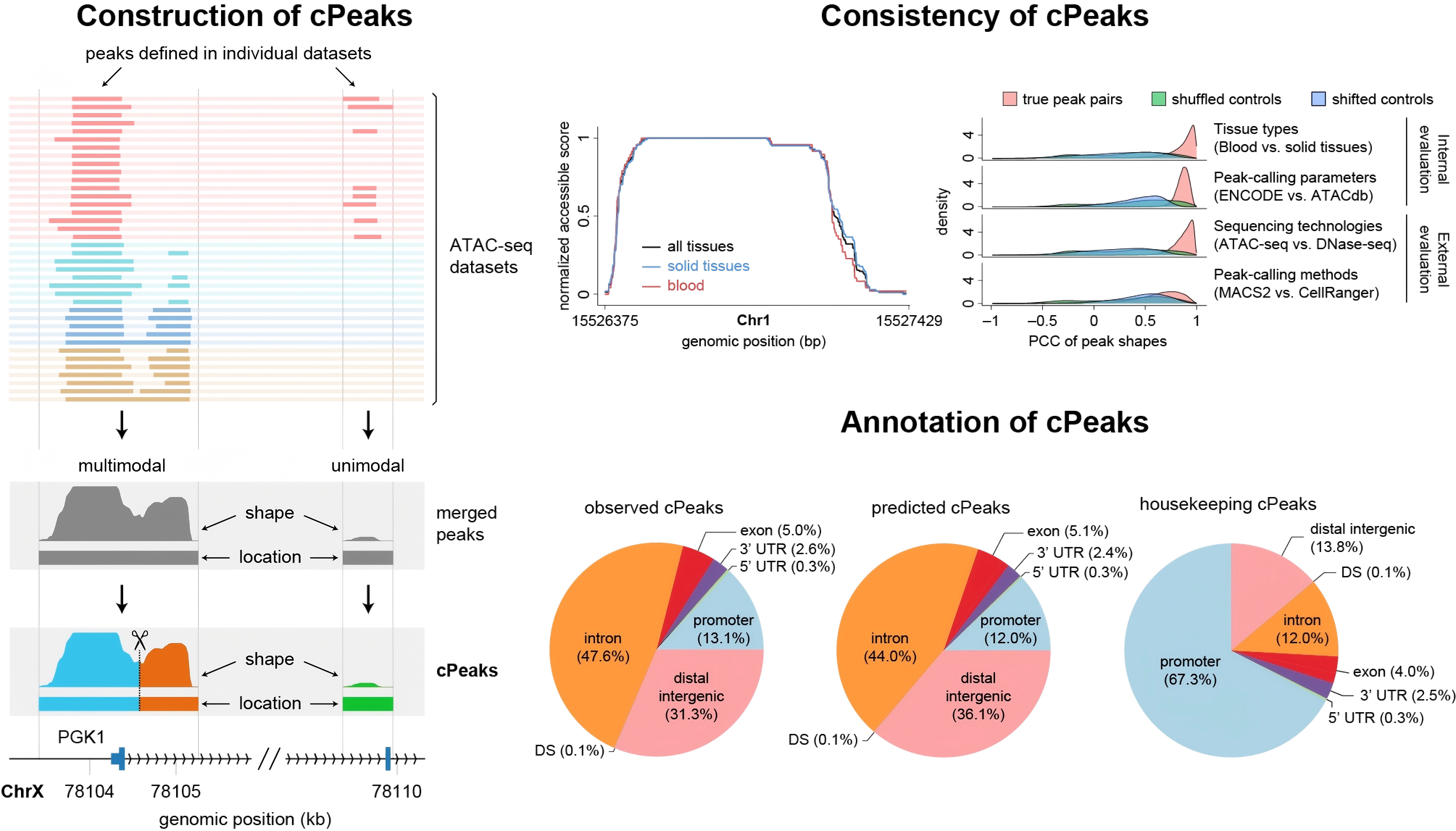
图1 cPeak的构建流程与性质
研究团队在多种不同数据集上系统比较cPeak与主流分析流程在单细胞聚类与注释任务中的表现。实验结果表明,在不同降维方法、不同特征数量与多种评价指标下,cPeak均稳定取得最优或接近最优的性能。这证明了cPeak可作为通用特征集合,为scATAC-seq数据分析提供稳健基础。
经典的pseudo-bulk peak calling方法常因细胞丰度较低而过滤掉罕见亚群的特异性峰。相比之下, cPeak的构建融合了广泛的组织类型,不被单一数据集中的细胞频率所主导,从而有效保留了区分低丰度细胞群体的关键信息。因此,cPeak展现出了对罕见细胞类型极高的鉴别敏感度。在人类外周血单核细胞数据分析中,cPeak成功且精准地鉴定出占比均不足1%多种罕见细胞亚群。是在针对pDC细胞的深度挖掘中,cPeak不仅鉴定出了数量远超传统pseudo-bulk peak和其他特征集的差异可及性区域,更是精准捕获到了pDC细胞标志基因启动子区域极具特异性的染色质可及信号(图2),充分体现了cPeak在解析罕见细胞类型方面的优势。

图2 cPeak在pDC中识别出更多特异性特征
研究发现,cPeak的内在的“形状”特征还蕴含调控机制信息。研究团队依据峰边缘长度与定位模式,将cPeak划分为精确定位(well-positioned)、不对称定位(asymmetrically-positioned)和弱定位(weakly-positioned)三种调控模式。其中,团队发现well-positioned cPeak的形成与先锋因子的结合以及侧翼核小体的精确定位密切相关。
团队深入探究了这类特异性峰在复杂细胞状态转换中的关键作用。例如,在人类胎儿视网膜发育数据分析中,well-positioned cPeak的比例在细胞谱系的过渡期显著上升,而在发育后期则出现回落(图3);在人类细胞图谱层面,胚胎细胞的well-positioned cPeak比例也普遍高于成体细胞,提示其在早期分化与发育过程中发挥重要作用;在妇科恶性肿瘤(子宫内膜癌与卵巢癌)数据中,基于cPeak特征的数据分析清晰区分了不同 CNV状态的肿瘤子克隆。随着肿瘤进展,well-positioned cPeak比例呈现非单调变化:早期至中期显著上升,而在晚期略有下降(图4)。该规律在独立肝细胞癌数据集中得到验证,表明 well-positioned cPeak的动态变化可能成为追踪肿瘤演化的重要信号。
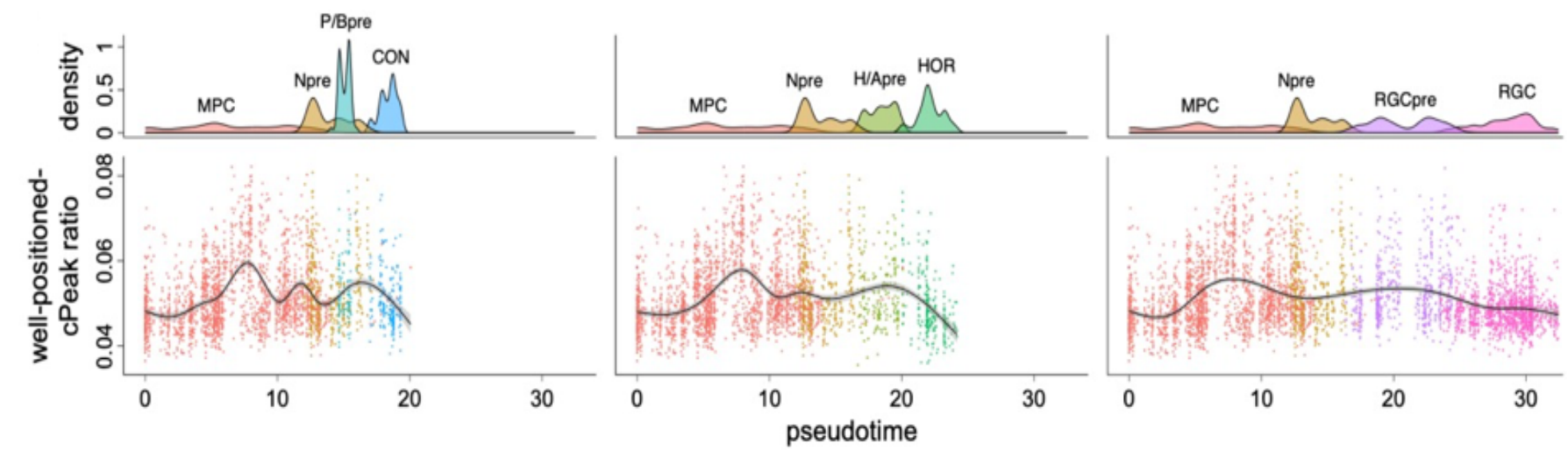
图3 发育过程中well-positioned cPeak比例的动态变化
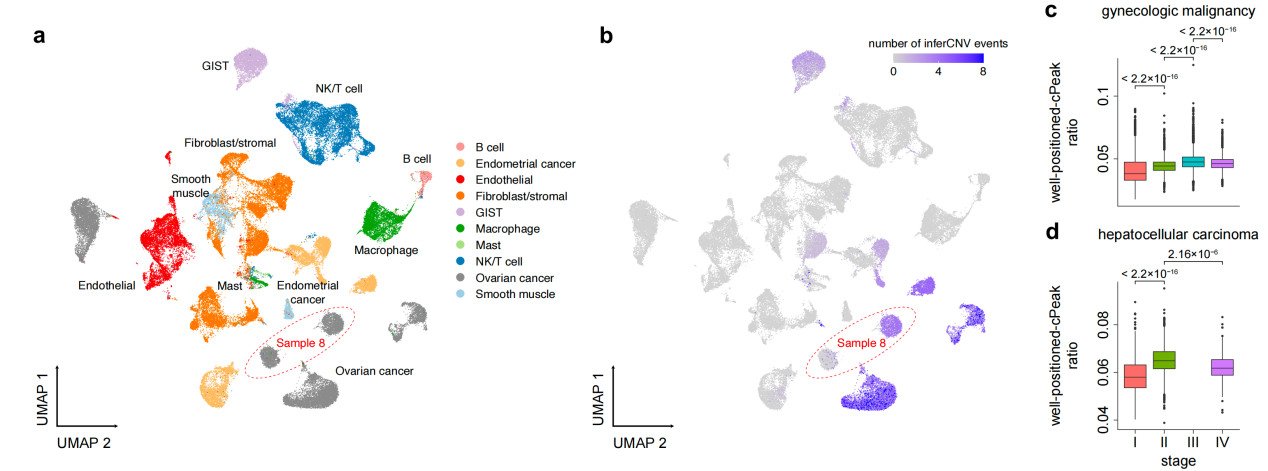
图4 cPeak在癌症数据分析中的表现
cPeak为scATAC-seq数据提供了统一、稳定且具生物学解释性的特征集合,显著简化分析流程,同时保留单细胞分辨率。研究结果表明,不同细胞类型虽在表型上高度异质,但在潜在染色质可及性层面共享一套基础特征结构。正如转录组研究依赖标准化基因注释体系,cPeak有望为表观组学建立统一特征字典,成为染色质可及性建模与跨数据集整合的重要底层基座,为未来多组学整合与表观组学大模型提供关键支撑。
清华大学自动化系博士生孟秋辰与硕士生吴鑫泽为该论文的共同第一作者;清华大学信息国家研究中心/自动化系张学工教授与清华大学信息国家研究中心魏磊助理研究员为本文的共同通讯作者。该研究得到了国家自然科学基金、科技部重点研发计划、清华–福州数据技术研究院的支持。
论文链接:Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun (2026). https://doi.org/10.1038/s41467-026-69461-6
供稿 | 数基生命系统交叉创新群体
编辑 | 李琳
审核 | 丁贵广 王钰言



